by Megan Amason
Think back to the last time you had a cold. What did you do to help alleviate your symptoms? Perhaps you took some medicine such as MucinexⓇ or TYLENOLⓇ, which are both examples of over-the-counter drugs. As the name implies, over-the-counter or OTC drugs are easily obtained at almost any store: all you have to do is find it on a shelf, purchase it at the register, and follow the instructions on the label. But what if you need something else in order to treat your cold, something that can’t be found on the shelf? What if you need a prescription drug, one that requires your doctor’s authorization to obtain and use? What makes prescription drugs different and how are they created? Part of the answer lies in the drug development process, or the way that scientists discover and test new medicines for treating various illnesses. (Read more on OTC vs prescription drugs here!)
What happens during the drug development process?
Broadly speaking, the purpose of the drug development process is to generate a way to treat an illness or disease, with the goal of either curing the illness or minimizing its symptoms. These treatments can be novel drugs, meaning they have not been tested or are being tested in a new combination, or the treatments can be previously approved drugs that are now being tested in a different illness than originally intended. This entire process is usually referred to as research and development, or R&D. As a way to protect public safety, this process is highly regulated by the U.S. Food and Drug Administration, or FDA, and can be broken down into five main steps (Figure 1). (Read about more responsibilities of the FDA here!)

Step 1: Discovery and Development
First, scientists identify an illness for which there are few or no treatments available, and then try to identify biological targets that can be potentially affected with a drug. Once a target is identified, scientists will work to develop drugs that can engage the target and lead to the desired outcome. In general, these drugs can originate from living organisms or they can be synthetically made, which is the key difference between biologics and small molecules, respectively. Regardless of its origin, once scientists find a drug that shows some promising results, this becomes their drug candidate, which moves on in the process for further investigations. Scientists often analyze thousands of compounds in their search for a good drug candidate! The pharmaceutical industry, which includes large companies such as Eli Lilly, Merck, and Pfizer, is generally responsible for the development of drugs, but drug development can also include universities or a collaboration between the two.
Before a drug candidate can be made into a medicine that is widely available, it must be rigorously tested. Of note, scientists must determine if the drug has the desired outcome and if the drug is safe for human use.
Step 2: Preclinical Research
Once a drug candidate is identified, there are many tests that must be performed before it can be used in people. The use of laboratory animals is essential in the drug development process because animals allow scientists to answer many important questions without involving people in potentially dangerous studies. While many insights can be gathered, there are three main categories of studies: pharmacokinetic (PK), pharmacodynamic (PD), and toxicology. In brief, here are the definitions and main questions that each type of study seeks to address:
- PK assesses how a drug moves throughout the body. A commonly used acronym for PK studies is ADME, which stands for absorption, distribution, metabolism, and excretion. These studies seek to understand how the body affects the drug.
- PD assesses the effects of a drug on the body. These studies seek to learn valuable information about the responses to different drug doses, as well as undesirable side effects.
- Toxicology, while similar to PK and PD studies, also encompasses studies that hope to identify any toxic or poisonous effects of a drug at various doses.
There are many different factors to consider when studying the effects of a drug, including age, sex, route of administration, and potential long-term effects, including increased risk for cancer or reproductive complications. As you can see, determining that a drug is safe for people is a huge task that takes time and must be performed with diligence.
Step 3: Clinical Development
Once preclinical studies are rigorously validated and the drug candidate is found to be both safe and effective in animals, an Investigational New Drug (IND) application must be filed with the FDA before studies can be conducted in people. The IND includes data gathered during preclinical studies, as well as information about the proposed clinical trials and a plan for the manufacturing of the drug. Compiling all of these data and plans is quite a daunting task, so many companies utilize the talents of medical writers to generate these applications (learn more about the exciting career of regulatory medical writing here!). After the IND is submitted, the FDA will take approximately 30 days to review the application; if no major issues are found, clinical trials may begin to recruit participants. It’s important to note that potential participants must undergo a process called informed consent where the drug is explained so that they can decide whether or not to participate. Because clinical trials are considered research studies, participants must understand the study design and be informed of the known and unknown risks of participating. Participants are informed that they can drop out of the trial at any time, for any reason. Briefly, there are three main phases of clinical trials, which are sequentially carried out and have different goals and purposes (Figure 2). Sometimes, there’s also a fourth phase (further discussed in Step 5 below).
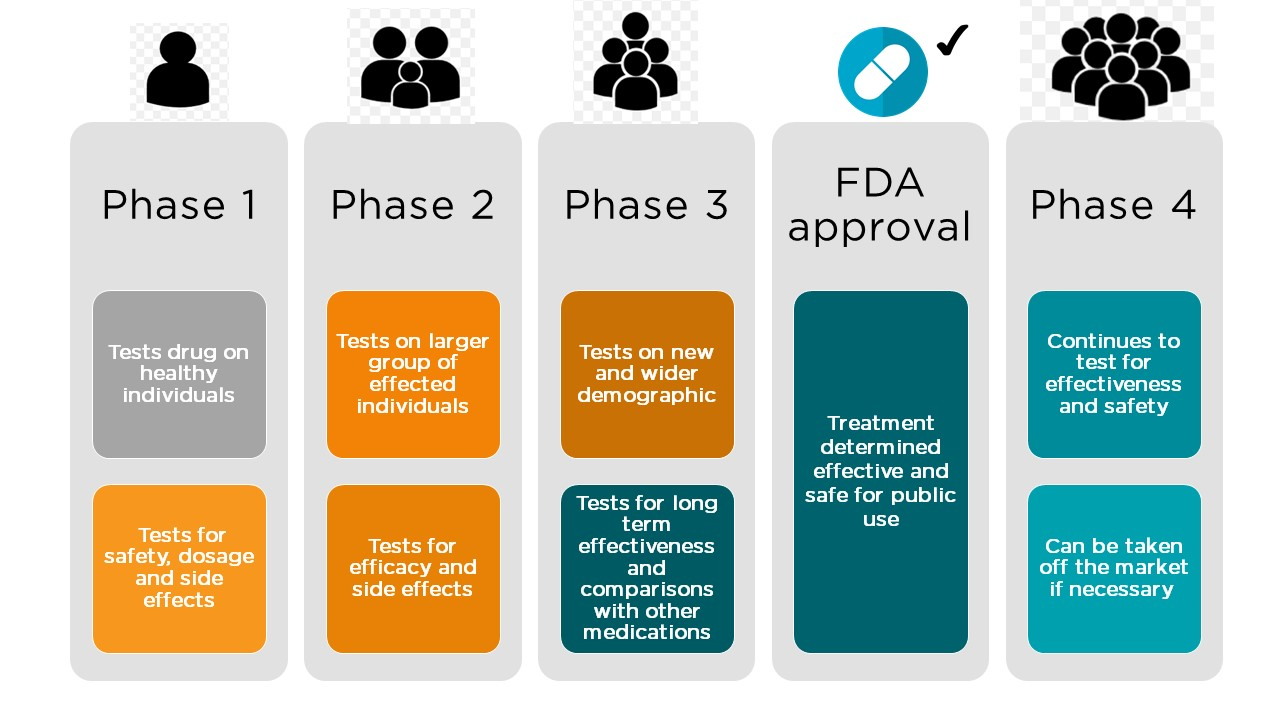
- Phase 1 clinical trials typically seek to answer one question: is the drug safe? Usually, phase 1 trials are conducted in less than a hundred healthy volunteers, with the exception of anti-cancer treatments. The purpose of phase 1 studies is to identify any negative side effects of the drug at various doses; in some cases, severe adverse events, or SAEs, can occur and must be reported to the FDA before clinical trials can resume.
- Phase 2 clinical trials involve individuals (usually a few hundred) with the specific illness, and seek to answer questions about the drug’s efficacy and effectiveness in generating desirable outcomes, such as improving a person’s symptoms or prolonging their life. Safety data are continually collected during this phase.
- Phase 3 clinical trials are similar to those in phase 2, but enroll even more study participants (usually in the thousands) to increase the power of the study to detect different effects of the drug treatment. Phase 3 studies also typically last longer, while still closely monitoring individuals for adverse reactions. However, an important exception is in the case of Orphan Drugs, or drugs that are designed to treat individuals with rare diseases. Overall study designs are altered to accommodate a lower number of participants, and companies will often be given specific incentives to develop these drugs.
It’s important to remember that trials can be stopped at any point for any reason. As new data are collected, decisions are made to reflect the new data which may alter initial plans made during preclinical studies. This is a good thing! In fact, the IND application is often considered a “living” document that is frequently updated, with updates being reported to the FDA at least annually. In total, a drug can be in clinical trials for 3 to 7 years, with only an estimated 30% completing all 3 phases and moving on to FDA review. As an extra way to safeguard potential participants and future consumers, applicable clinical trials must be registered at ClinicalTrials.gov, and the results made publicly available.
Step 4: FDA Review
At the completion of all clinical trials, if there’s evidence that the drug has been safe and effective in treating the illness, all of the data generated from both nonclinical and clinical studies must be compiled into a New Drug Application, or NDA. The primary purpose of an NDA is to provide the FDA with all of the information necessary for them to determine if the drug should be widely available for individuals. As you can imagine, there is a lot of information to consider, so the review process can take quite some time: anywhere from 6 to 12 months! During this time, the FDA can request more information or even ask for further studies to be conducted. However, in some cases investigators can file for a Fast Track review if the drug shows promise in treating a serious condition that has few treatment options, which is usually the case with certain kinds of cancer.
Not every NDA is approved, but if the FDA decides that a drug is safe and effective and it is approved, there is still a lot of work that needs to be done before doctors can begin prescribing the new drug.
Step 5: Post-Market Monitoring
An important step that occurs immediately after NDA approval is called labeling. This is where the FDA and investigators determine the information that must be printed on the drug container, as well as the information that must be supplied to doctors and pharmacists who will prescribe and distribute the drug. This information is critical in educating doctors, pharmacists, and patients on the intended use of the drug, how to take the drug, and any side effects that could occur. Once a new drug is finally on the market, this doesn’t mean that the journey is over. In fact, post-approval monitoring, sometimes referred to as phase 4 clinical trials, are an important part of the continued surveillance to make sure the drug is safe. As long as a drug is on the market, the FDA requires safety monitoring which allows doctors, pharmacists, and consumers to report any issues with the drug. Of particular importance is the reporting of any rare side effects that were not observed until more individuals started taking the drug after its approval. While uncommon, drugs can be withdrawn from the market if reported side effects are serious and occur frequently enough to negate the positive effects of the drug.
A lengthy, and costly, process
Getting a drug to market is no small feat and takes the cooperative efforts of an incredibly large group of people. It’s estimated that the entire drug development process can take anywhere from 12 to 15 years, and can cost up to $1 billion for a single drug! Why is this the case? Are there ways to improve the drug development process? How can we work towards developing drugs more quickly, while cutting the cost of drugs for patients who desperately need them?
One of the issues lies in the fact that preclinical studies, aimed at assessing the safety of the drug, are rarely predictive of clinical success or efficacy. This means that there is already an incredible amount of time and resources invested in a drug before significant evidence supports its effectiveness. Even worse, if a drug candidate proceeds all the way to phase 3 trials before it’s determined unsafe or ineffective, a lot of time and money were essentially wasted on a drug that cannot be sold. Therefore, one way that the process can be improved is by identifying biomarkers, or measurable characteristics that may be more indicative of efficacy even during preclinical studies. Biomarkers can serve many purposes, but by gathering these safety and efficacy data earlier on, scientists can save valuable time and money. Of course, while improvements are definitely needed, any changes to the drug development process should be made without compromising the safety of individuals.
The next time your doctor prescribes you medicine, hopefully you can appreciate just how much time, effort, and money were invested in developing and testing that drug before it could help you!
