by Jenna Grabowski
When people hear the term “infectious disease,” they typically think of diseases caused by viruses, bacteria, or parasites; not often do diseases caused by prions come to mind.
What is a prion?
To understand what a prion is, it is first important to understand proteins. Proteins are a class of molecules that are made up of amino acids. Our DNA encodes messages that are transcribed into RNA. These RNA messages are then used to code for amino acids that build a protein. The RNA tells the protein building machine (the ribosome) what amino acids to put in order. These amino acids create a long chain that folds into a unique shape characteristic of that protein. This correctly folded protein can go about the cell and perform its necessary functions.

Figure describing how messages are transcribed (from DNA to RNA) and translated (from RNA to amino acids) to create working proteins. Proteins fold from a polypeptide chain into a working protein. Made with BioRender.
Throughout this process of building a protein, many things can go wrong. Mutations or defects in the DNA/RNA sequence can cause a protein to lose an amino acid or change the amino acid to a different one (which can either have a bad consequence for the protein or no consequence at all). Proteins can also have problems folding (despite having no problems in the DNA or RNA). They can fold into an incorrect shape or not fold at all. While the cell has mechanisms to prevent or fix this, they can sometimes fail and allow the misfolded protein to cause damage. Once a protein is misfolded, it loses its ability to perform its regular function. It can also influence other proteins. When a specific protein, known as the prion protein (or PrP for Prion Protein), has trouble folding, it sticks to other prion proteins and causes them to misfold as well. These proteins get stuck together and aggregate, or form clumps, and—voilà—a prion disease has occurred.
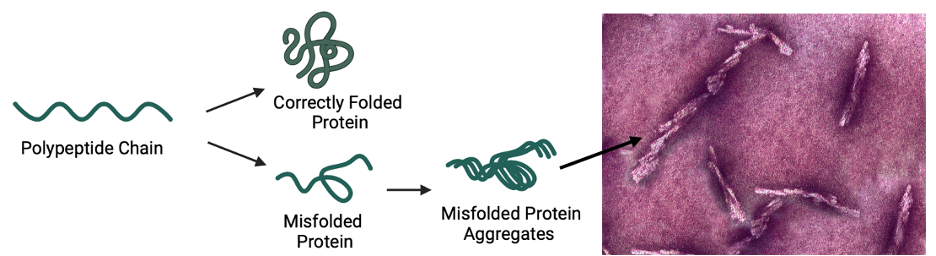
(Left) Figure describing how proteins can become prion clumps. Made with BioRender.
(Right) Image of infectious prion protein aggregates under a microscope.
How do prions cause disease?
We all have the prion protein (PrP) in our normal cells and tissue. If the protein is healthy and correctly folded it does not cause any negative effects. This healthy form of the prion protein is known as PrPC (normal cellular prion protein). However, once the prion protein (PrP) becomes misfolded, it can cause complications. This misfolded and pathogenic version of the prion protein is known as PrPsc (standing for scrapie prion protein). The PrPsc sticks to other PrPc proteins and causes them to misfold as well, turning them into the PrPsc form. This leads to aggregates, or clumps, of misfolded prion proteins that can cause damage to organs. Clumps of misfolded proteins get in the way of regular cellular function and cause problems in the cell. The prion protein is largely found in the brain; therefore, when this protein begins to aggregate, brain damage occurs which leads to the disease symptoms such as memory impairment, personality changes, and difficulty moving. Prion diseases are also known as transmissible spongiform encephalopathies (TSEs) due to the tiny holes in the brain (caused by protein aggregates) that make the brain look “spongy” under a microscope.
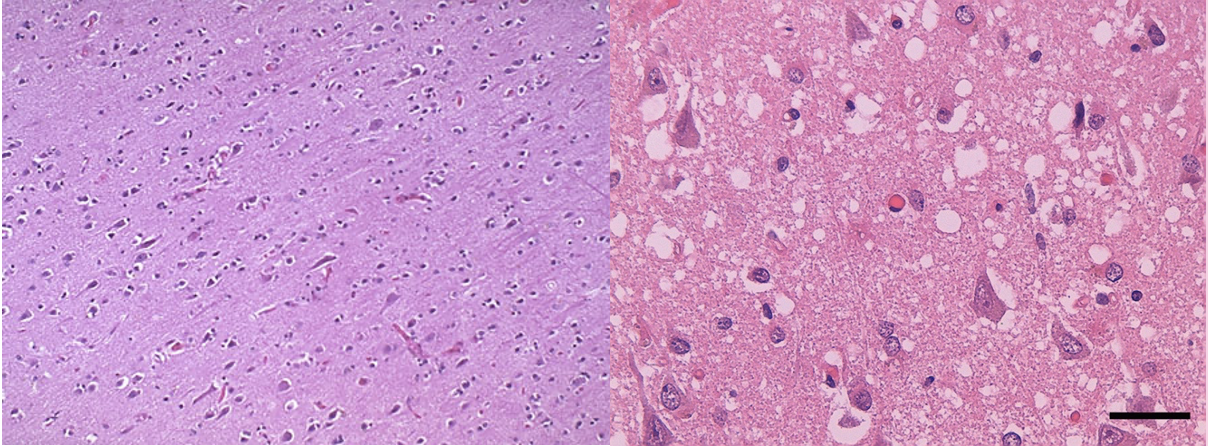
Left: microscope image of normal, healthy brain tissue.
Right: microscope image of brain tissue from a patient who had Creutzfeldt-Jakob Disease; notice all the holes in the tissue that give the brain a spongy appearance.
What are examples of prion diseases?
Prion diseases include both known human and animal diseases. Bovine Spongiform Encephalopathy (BSE), more commonly known as Mad Cow Disease, is a prion disease found in cattle. Scrapie is a prion disease found in sheep. Chronic Wasting Disease (CWD) is a prion disease found in deer, moose, and elk. Human prion diseases include Creutzfeldt-Jakob Disease (CJD), Fatal Familial Insomnia, and the first human prion disease discovered, Kuru.

Image: Chronic Wasting Disease (CWD) is a prion disease that affects deer, moose, and elk.
How does someone get a prion disease?
Prion diseases are infectious diseases, meaning they can be spread between organisms. The spread of prions is mediated through exposure to contaminated tissue. Examples of this can be found in the history of Kuru, in which the Fore people of New Guinea practiced ritualistic cannibalism in which relatives ate tissues (including prion infected brains) of deceased family members. In nature, a wild animal can eat the tissue remains of a deceased prion-infected animal. Consuming this infected tissue can lead to an organism developing a prion disease.
Prion disease can also occur without exposure to an already infected organism. This can happen in two different ways. First, people can develop random mutations in their DNA that lead to the misfolding of the PrPC into the PrPsc form, which will then lead to development of a prion disease. This is usually associated with aging. Another way to develop a prion disease is through inheriting this already mutated DNA from your family. These two methods differ from the first as someone is born with the susceptibility to a prion disease, rather than acquiring an infection during their lifetime through exposure to another infected organism.
Prions are a unique form of infectious disease that push us to redefine our boundaries and knowledge in biology. Further research will hopefully allow us to gain a better understanding of how these diseases occur, how to treat and prevent them, and what other infectious particles are out there.
