by Hazel Milla
Fun Rating: 4/5

Difficulty Rating: 4/5
What is the general purpose?
We can use microarray technology to measure DNA methylation levels in an organism’s DNA. By doing so, we determine how biology and environment influence gene expression.
Why do we use it?
DNA methylation is a type of epigenetic modification, or in other words, a chemical change to the genome that does not alter the underlying sequence of DNA. Specifically, methylation is the addition of a methyl group to a cytosine nucleotide. This chemical change to the nucleotide makes the DNA less accessible to proteins that transcribe it into RNA, preventing a gene from being expressed as a protein. Measures of DNA methylation and other epigenetic modifications can indicate how an organism’s biology and environment affect gene expression. This information can give us insight into how various factors contribute to the development of disease. For example, we can identify epigenetic changes associated with smoking to further our understanding of how smoking leads to negative health outcomes. DNA methylation microarrays can also be used for disease prediction and diagnostic purposes.
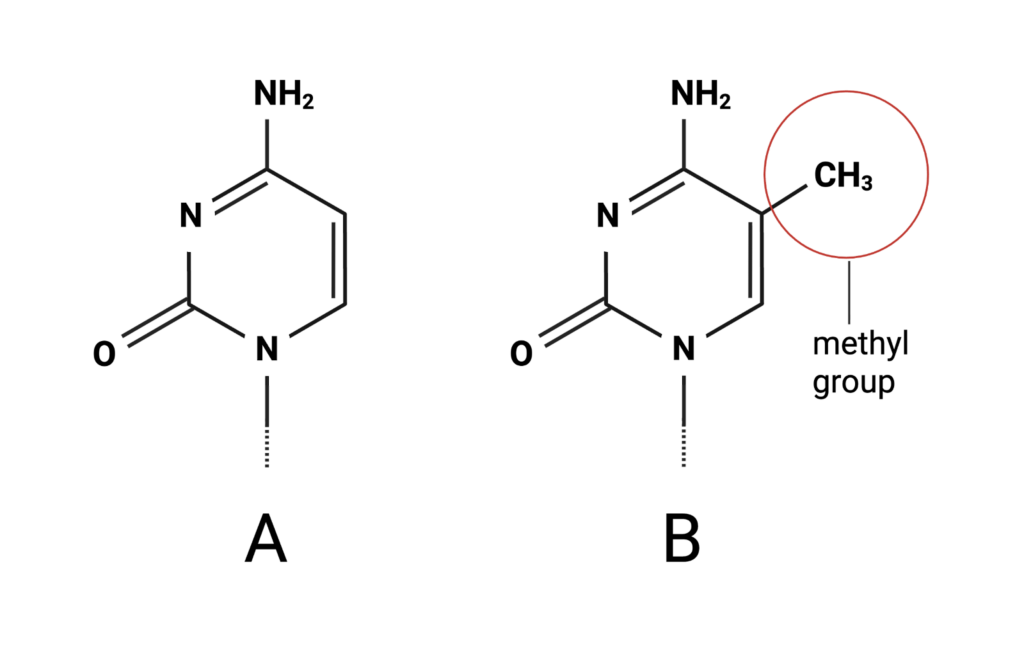
Figure 1. A cytosine structure with a methyl group attached. Figure created by author in Biorender.
How does it work?
DNA methylation microarrays work through two primary steps: data collection and analysis.
Data collection
First, a tissue sample is collected for analysis. Blood, tumors, skin, and the brain can be analyzed with this technology, among other tissue types.
After collecting the tissue sample, double-stranded DNA is isolated from the rest of the sample through DNA extraction and split apart through denaturation. The single-stranded DNA is then chemically treated to label which sites are methylated vs unmethylated.
How is the DNA chemically treated?
DNA consists of four types of nucleotides: cytosine (C), guanine (G), adenosine (A), and thymine (T). Cytosines followed by guanine, referred to as CpG sites, are where DNA strands can become methylated. To determine if a cytosine at a CpG site is methylated, the DNA is treated with a chemical mixture called sodium bisulfite. The ensuing reaction, bisulfite conversion, causes unmethylated cytosines to be converted to uracil, another type of nucleotide that is present in RNA. These uracils are then recognized as thymine during later processing. However, if the CpG is methylated, the cytosine is immune to this chemical reaction and remains unchanged. To determine if a cytosine at a specific CpG site has been converted, we need to assess the underlying DNA sequence following bisulfite treatment.
How is the DNA sequence determined after bisulfite conversion?
Millions of copies of the converted DNA are generated through polymerase chain reaction (PCR). The DNA is then added to an array, a plate consisting of small wells lined up in rows and columns. Each well contains silicon beads covered with probes. These probes are single-stranded DNA pieces that will bind, or “hybridize,” to up to 937,690 DNA sequences simultaneously. Following hybridization, probes are stained with fluorescent chemical compounds called fluorophores, which light up when exposed to a laser. Probes detect whether or not the target DNA has been converted and bind, emitting fluorescent signals accordingly. The light emitted by these probes is measured using a detector.
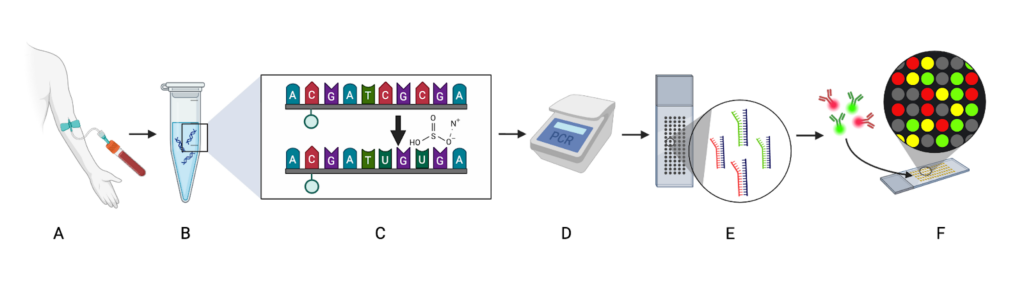
Figure 2. After collecting a blood sample (A), DNA is extracted (B), denatured, and treated with bisulfite (C), which converts unmethylated cytosines to uracils. After PCR amplification (D), which converts the uracils to thymines, the DNA hybridizes to probes on the microarray (E). The probes are stained with fluorophores that emit light when exposed to a laser (F). Figure created by author in Biorender.
Data processing and analysis
The light emitted by the probes needs to be cleaned up and converted into data that can be analyzed. To do this, we use software to filter out noisy signals and convert true signals to a value between 0 and 1, called a beta value. A beta value of 0 at a CpG site indicates no methylation, while a value of 1 indicates complete methylation, and anything between means that partial methylation occurred.
After processing the data, the resulting beta values can be used to analyze the relationship between methylation levels at specific genes and environmental factors such as pollution exposure, or biological factors such as age. Linear regression is a common method used to analyze the relationship between DNA methylation levels and a variety of factors.

Figure 3. Researchers use computational methods to process and analyze the data. Figure created by author in Biorender.
DNA methylation can shape how a cell differentiates into specialized cell types. It can change as a healthy response to biological and environmental factors, or be altered in ways that contribute to disease risk. However, epigenetic changes are potentially reversible, making them a promising candidate for disease treatment and prevention. Microarrays are a common, effective way to study DNA methylation to better understand biology and to diagnose, predict, treat, and prevent disease.
