by Hailey Dodson
Cystic Fibrosis (CF) is a well-known genetic disease most often associated with severe lung problems. For decades, it has been framed primarily as a respiratory condition because the associated complications in the lungs have been the leading cause of illness and early death.
CF is caused by recessive mutations in the cystic fibrosis transmembrane conductance receptor (CFTR) gene, which encodes an ion channel protein that helps regulate the hydration of mucus (Figure 1). When CFTR is dysfunctional, the mucus becomes thick and sticky, leading to progressive lung damage and chronic issues such as shortness of breath, persistent cough, and recurring infections. Given the severity of these symptoms, it is no surprise that research and clinical management have historically centered on the lungs. However, CFTR is important and active throughout the body, and its dysfunction impacts nearly all mucus-producing organs, including the gastrointestinal (GI) tract, pancreas, liver, and reproductive tract.
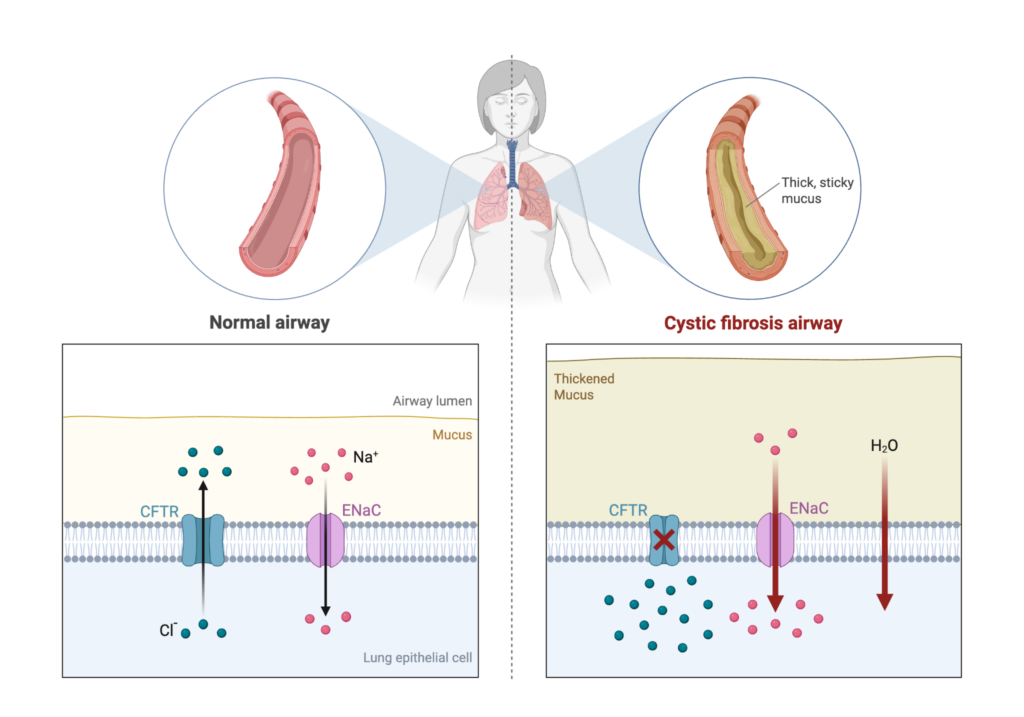
Figure 1. A look at cystic fibrosis in the lungs. The CF lung is characterized by changes to airway structure and the mucus layer, resulting in the constriction of the airways, leaving them vulnerable to obstruction and recurrent infection. Image created by the author using BioRender.
Over the past decade, scientists have developed new CFTR-modulating therapies that have relieved respiratory symptoms for many people with CF, improving both their quality of life and life expectancy. As lung health has improved, researchers and clinicians have begun to focus their attention on the symptoms caused by other affected organs, especially the GI system.
GI symptoms vary widely between individuals with CF. Some people experience little to no digestive discomfort, while others face persistent issues such as constipation, greasy stools, cramping, and bloating (Figure 2). One of the earliest signs of CF in newborns is meconium ileus, a life-threatening bowel obstruction caused by thickened mucus. Even if individuals do not display early GI problems, they remain at risk for developing significant symptoms later into adulthood. Beyond the direct effects on the intestines, GI dysfunction can have broader complications, including poor nutrient absorption resulting in poor growth and failure to thrive, and an increased risk of colon cancer. These challenges highlight why CF’s impact on patient life spans much further beyond the lungs.
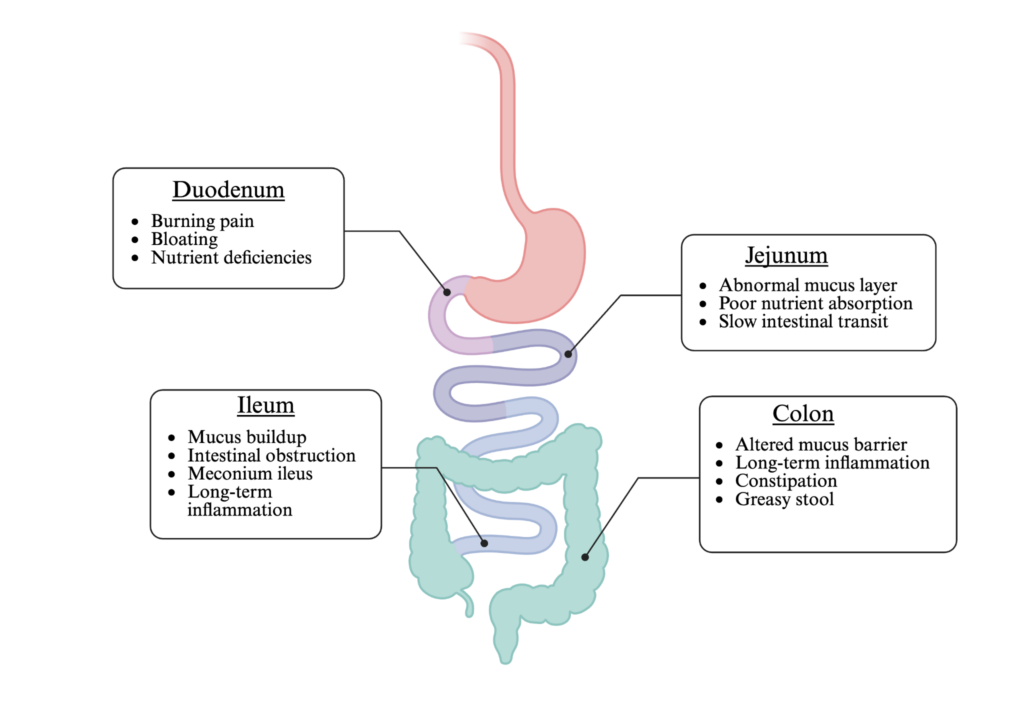
Figure 2. CF-associated complications along the gastrointestinal tract. CFTR dysfunction leads to a variety of manifestations along the different regions of the gastrointestinal tract. Image created by the author using BioRender.
Despite the seriousness of GI-associated complications in CF, our knowledge about the mechanisms behind them remains limited. This is partially due to significant limitations of the current model systems available for studying the CF gut. CF mouse models are unable to accurately replicate the human microbiome and lack key cell populations that are found in the human intestinal lining. To overcome these limitations, researchers are turning to the use of new human-oriented model systems. Among the most promising is the use of human intestinal organoids, lab-grown organs designed to mimic the intestinal environment. These organoids can be developed to model any region of the GI tract and exhibit all the required intestinal cell types. Importantly, goblet cells, which are responsible for secreting mucus into the intestine, secrete mucus into the center of the organoid, allowing researchers to collect and analyze mucus directly. Models like this are helping to push the field of CF research beyond the lungs and into the gut.
